什么是亞瑟綜合癥?
Usher綜合癥是影響聽力和視力的最常見病癥; 有時它也會影響平衡���。Usher綜合癥的主要癥狀是耳聾或聽力喪失以及稱為視網(wǎng)膜色素變性(RP)的眼病[re-tin-EYE-tis pig-men-TOE-sa]����。

Usher綜合癥的耳聾或聽力喪失是由內(nèi)耳毛細(xì)胞(聲音受體細(xì)胞)的異常發(fā)育引起的�����。大多數(shù)患有亞瑟綜合癥的兒童出生時伴有中度到嚴(yán)重的聽力損失,具體取決于類型���。不太常見的是�����,Usher綜合癥的聽力損失出現(xiàn)在青春期或以后�。Usher綜合癥還可能由于前庭毛細(xì)胞的異常發(fā)育�����,檢測重力和頭部運(yùn)動的感覺細(xì)胞而導(dǎo)致嚴(yán)重的平衡問題�����。
RP最初通過視網(wǎng)膜細(xì)胞的進(jìn)行性退化導(dǎo)致夜盲和外周(側(cè))視力喪失���。視網(wǎng)膜是眼睛后部的光敏組織,對視力至關(guān)重要�。隨著RP的進(jìn)展,視野變窄��,直到只有中心視力仍然存在,這種情況稱為隧道視覺��。黃斑中的囊腫[MAC-u-la](視網(wǎng)膜的中央部分)和白內(nèi)障(晶狀體混濁)有時會導(dǎo)致Usher綜合癥患者中央視力的早期下降��。
誰受到Usher綜合癥的影響?
Usher綜合癥發(fā)病率約為4到每10萬人17人���,占所有遺傳性耳聾��,失明病例約50%�����。這種情況被認(rèn)為占所有聾兒的3%至6%�,另有3%至6%的兒童難以聽力���。
什么原因?qū)е聛喩C合癥?
Usher綜合癥是遺傳的����,這意味著它通過基因從父母傳給孩子��。每個人遺傳一個基因的兩個拷貝��,每個父母一個�����。有時基因會被改變或發(fā)生變異。突變基因可能導(dǎo)致細(xì)胞發(fā)育或異常��。
Usher綜合癥是一種常染色體隱性遺傳病�。“常染色體”意味著男性和女性同樣可能患有這種疾病����,同樣可能將其傳給任何性別的孩子?����!半[性”意味著只有當(dāng)一個孩子繼承了同一個錯誤基因的兩個拷貝時才會發(fā)生這種情況����,每個父母一個。具有一個異常Usher基因的人沒有這種疾病���,但是有50%的機(jī)會將異?;騻鬟f給每個孩子��。當(dāng)具有相同突變Usher綜合癥基因的兩個攜帶者有一個孩子在一起時��,每個出生有一個:
有四分之一的機(jī)會讓孩子既沒有亞瑟綜合癥����,也沒有攜帶者。
有四分之二的機(jī)會讓孩子成為不受影響的運(yùn)營商�。
有一個孩子患有亞瑟綜合癥的機(jī)會有四分之一。
三種類型的Usher綜合癥有哪些特征?
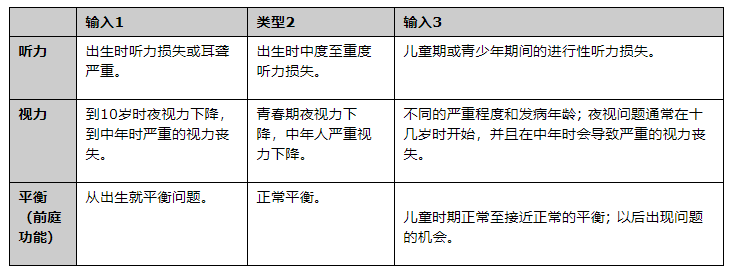
Usher綜合癥有三種類型�����。在美國�����,類型1和類型2是最常見的����。它們共同占Usher綜合癥病例的95%。
1型:1型Usher綜合癥患兒出生時有嚴(yán)重的聽力損失或耳聾�,并有嚴(yán)重的平衡問題。許多人從助聽器中獲得很少或沒有獲益��,但可能是人工耳蝸的候選者 - 這種電子設(shè)備可以為嚴(yán)重聽力喪失或耳聾的人提供聲音感�����。家長應(yīng)盡早咨詢孩子的醫(yī)生和其他聽力保健專業(yè)人員,以確定孩子的溝通方式���。當(dāng)大腦最容易接受學(xué)習(xí)語言時�,無論是口語還是簽名�,都應(yīng)該及時開始干預(yù)。
與1型Usher綜合癥相關(guān)的平衡問題在沒有支持的情況下延遲坐位�����。步行很少發(fā)生在18個月之前��。1型Usher綜合癥的視力問題通常在10歲之前開始����,從難以在夜間看到并且在幾十年內(nèi)發(fā)展為嚴(yán)重視??力喪失開始。
2型:2型Usher綜合癥患兒出生時伴有中度至重度聽力損失但平衡正常����。盡管聽力損失的嚴(yán)重程度各不相同,但大多數(shù)患有2型Usher綜合癥的兒童可以口頭交流并從助聽器中受益�。RP通常在2型Usher綜合癥患者的青春期后期診斷。
3型:3型Usher綜合癥患兒出生時聽力正常�。大多數(shù)人的平衡接近正常,接近正常�,但有些人隨著年齡的增長而出現(xiàn)平衡問 聽力和視力的下降各不相同��?�;加?型Usher綜合癥的兒童經(jīng)常在青春期發(fā)生聽力損失,需要在成年中后期使用助聽器��。夜盲也通常在青春期開始���。在十幾歲到二十出頭的時候出現(xiàn)了盲點(diǎn)��。法律失明經(jīng)常發(fā)生在中年��。
亞瑟綜合癥如何診斷?
亞瑟綜合癥的診斷涉及有關(guān)該人的病史和聽力���,平衡和視力測試的相關(guān)問題。早期診斷很重要��,因?yàn)樗梢蕴岣咧委煶晒β?���。眼保健專家可以使用擴(kuò)張液來檢查視網(wǎng)膜是否有RP的跡象。視野測試測量周邊視力���。視網(wǎng)膜電圖[e-lec-tro-RET-in-o-gram]測量視網(wǎng)膜中眼睛的光敏細(xì)胞的電響應(yīng)����。光學(xué)相干斷層掃描可能有助于評估黃斑囊性變化。Videonystagmography [vi-de-o-nigh-stag-MAH-gra-fee]測量無意識的眼球運(yùn)動��,這可能意味著平衡問題���。聽力學(xué)測試確定一系列頻率的聽力靈敏度�。
基因檢測可能有助于診斷亞瑟綜合癥���。到目前為止��,研究人員已經(jīng)發(fā)現(xiàn)了導(dǎo)致亞瑟綜合癥的9個基因�����。所有人都可以進(jìn)行基因檢測:
1型Usher綜合癥:MY07A�����,USH1C�,CDH23���,PCHD15����,USH1G
2型Usher綜合癥:USH2A,GPR98�,DFNB31
3型Usher綜合癥:CLRN1
Usher綜合癥的基因檢測可通過臨床研究獲得。
亞瑟綜合癥如何治療?
目前�����,Usher綜合癥尚無法治愈��。治療涉及管理聽力���,視力和平衡問題。早期診斷有助于定制教育計劃��,考慮聽力和視力喪失的嚴(yán)重程度以及兒童的年齡和能力�����。治療和通信服務(wù)可包括助聽器����,輔助聽力設(shè)備,人工耳蝸植入���,聽覺(聽力)訓(xùn)練和/或?qū)W習(xí)美國手語���。獨(dú)立生活培訓(xùn)可能包括平衡問題的定向和流動培訓(xùn)�,盲文教學(xué)和低視力服務(wù)�。
根據(jù)國家眼科研究所和基金會抗擊失明基金會支持的長期臨床試驗(yàn)結(jié)果,維生素A可能會減緩RP的進(jìn)展����。根據(jù)該研究,患有常見RP形式的成人可以從每日補(bǔ)充15,000 IU(國際單位)的棕櫚酸形式的維生素A中受益����。患者應(yīng)該在繼續(xù)之前與他們的醫(yī)療保健提供者討論這種治療方案�����。由于1型Usher綜合癥患者未參加本研究����,因此不建議對這些患者使用高劑量維生素A.
維生素A補(bǔ)充劑的一般預(yù)防措施:
不要用β-胡蘿卜素補(bǔ)充劑替代維生素A棕櫚酸酯。
不要服用大于推薦劑量15,000 IU的維生素A補(bǔ)充劑或改變飲食以選擇含有高水平維生素A的食物�����。
由于出生缺陷風(fēng)險增加,孕婦不應(yīng)服用高劑量維生素A補(bǔ)充劑����。考慮懷孕的女性應(yīng)該在嘗試懷孕前停止服用大劑量維生素A補(bǔ)充劑六個月��。
對Usher綜合癥進(jìn)行了哪些研究?
研究人員正試圖找出導(dǎo)致亞瑟綜合癥的其他基因�。努力將導(dǎo)致改善遺傳咨詢和早期診斷,并可能最終擴(kuò)大治療選擇��。
科學(xué)家們也在開發(fā)具有類似于Usher綜合癥特征的小鼠模型�。使用小鼠模型進(jìn)行的研究將有助于確定亞瑟基因的功能并為潛在的治療提供信息�����。
其他研究領(lǐng)域包括開發(fā)新方法����,用于早期識別患有Usher綜合癥的兒童,改善使用助聽器或人工耳蝸植入聽力損失的兒童的治療策略����,以及測試創(chuàng)新干預(yù)策略以幫助減緩或阻止RP的進(jìn)展。臨床研究人員也在描述具有各種類型的Usher綜合癥的個體之間的平衡變化。
更多詳情請登錄聽覺有道助聽器www.xzjxt.cn